
ライソゾーム病に関して(各論)医師向けムコ多糖症
1.概要、欠損酵素
ムコ多糖症は7つの病型を含む疾患群の総称である。体内のムコ多糖を分解するライソゾーム酵素が欠損することにより、全身にムコ多糖が蓄積し、骨関節病変、皮膚・結合組織病変、中枢神経障害、呼吸器・循環器・消化器など全身性の多様な臨床所見を呈する。
表1にムコ多糖症の分類を示す。それぞれの病型で重症型から軽症型まで幅広い臨床的重症度の患者が存在する。
| 病型 | 遺伝形式 | 欠損酵素 | 尿中MPS | |
|---|---|---|---|---|
| I | Hurler Hurler/Scheie Scheie |
AR | α-L-iduronidase | DS, HS |
| II | Hunter | XR | Iduronate-2-sulfatase | DS, HS |
| III | Sanfillipo A B C D |
AR | Heparan-N-sulfatase α-N-acetylglucosaminidase acetyl-CoA:α-glucosaminide N-acetyltransferase N-acetyl-glucosamine-6-sulfatase |
HS |
| IV | Morquio A B |
AR | N-acetylgalactosamine-6-sulfatase β-galactosidase |
KS, CS |
| VI | Maroteau-Lamy | AR | N-acetylgalactosamine-4-sulfatase | DS |
| VII | Sly | AR | β-glucuronidase | DS, CS |
| IX | AR | hyaluronidase | HA | |
| AR:常染色体劣性遺伝、XR:X染色体連鎖劣性遺伝 DS:デルマタン硫酸、HS:ヘパラン硫酸、KS:ケラタン硫酸、CSコンドロイチン硫酸 HA:ヒアルロン酸 |
||||
2.遺伝形式
Hunter症候群(X染色体連鎖劣性遺伝)以外は常染色体劣性遺伝形式をとる。
3.分類
I型は重症の Hurler 症候群、中等症の Hurler/Scheie、軽症の Scheie 症候群に分類される。II型(Hunter 症候群)は中枢神経障害を伴う重症型と、中枢神経障害を伴わない軽症型に大別されるが、種々の中間的重症度の患者が存在する。III型(Sanfillipo症候群)は原因酵素の異なる4亜型(A, B, C, D)が存在し、臨床的にはいずれも類似の所見を呈する。IV型(Morquio症候群)は本来のムコ多糖症であるA型と、β-galactosidase の欠損を原因とするB型に分類される。VI型、VII型も種々の重症度が存在する。IX型は1例報告されているのみである。V型、VIII型は欠番となっている。
4.人種差、発症頻度
欧米ではI型が多いが、日本、韓国などの東アジアではII型が過半数を占め、次いでIII型、I型の順になっている。発症頻度は欧米では24,000人に1名、本邦では5~6万人に1名と推定されている。

5.症状
ムコ多糖症に共通する症状としては、関節拘縮、骨格変形、低身長、特徴的顔貌、巨舌、厚い皮膚、多毛、気道狭窄、反復性呼吸器感染、難聴、心臓弁膜症、肝脾腫、臍・そけいヘルニア、中枢神経障害などがある。病型別には以下のような特徴がある。
- I型(Hurler/Scheie症候群)
- 重症の Hurler 症候群は乳児期から特徴的顔貌,関節拘縮,椎体変形、精神運動発達遅滞、角膜混濁、肝脾腫、ヘルニアなどを認め、呼吸不全、心不全などで小児期に死亡する。軽症の Scheie 症候群では知能はほぼ正常で成人に達し、社会生活を送れる例もある。
- II型(Hunter 症候群)
- Hurler 症候群に類似した症状であるが、角膜混濁はなく、皮膚に特徴的丘疹を認めることが多い。重症型は幼児期から精神運動発達遅滞、多動を認め、学童期以降に退行し、小児期に死亡する。軽症型は幼児期に発症し、知能はほぼ正常で、社会生活を送れる例もある。
- III型(Sanfilippo 症候群)
- 精神運動発達遅滞、行動異常(睡眠障害、多動、粗暴)、難聴、痙攣、退行などの神経症状が主体で、骨格異常、肝脾腫などの身体症状は軽いが、粗な毛髪、多毛は特徴的である
- IV型(Morquio 症候群)
- 脊椎後彎・側彎、短胴型低身長、Ⅹ脚、関節過伸展、環軸椎亜脱臼などの骨格病変が強く、角膜混濁、難聴、弁膜症なども認められるが、知能は正常である。頸椎病変の程度が身体障害と予後を大きく左右する。
- VI型(Maroteaux-Lamy 症候群)
- Hurler 症候群類似の身体所見を認めるが知能は正常である。
- VII型(Sly 病)
- Huler 症候群や Morquio 症候群に類似の所見を呈する。
6.診断
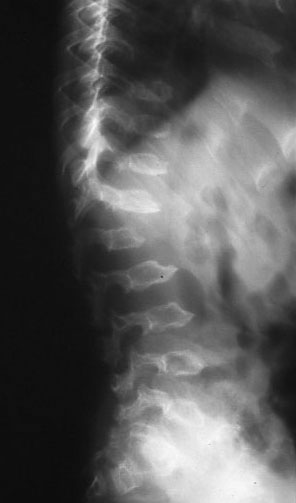
上記の臨床所見に加え、特徴的骨レントゲン所見(dysostosis multiplex)が診断の手がかりとなる。大きく厚い頭蓋骨とトルコ鞍の変形(J-shape, ballooning)環軸椎の低形成・亜脱臼、胸腰部椎体の卵形化・扁平化、肋骨の扁平化(ボートのオール様)、長管骨端の異形成、手根骨低形成、指骨の短縮(砲弾形)などが参考になる。尿中ムコ多糖の排泄増加と、それぞれの病型に特徴的な排泄パターン(表1)によってスクリーニングし、酵素活性の測定により確定診断される。
保因者診断、出生前診断では遺伝子解析が必要となる場合も多い。
図2 特徴的なレントゲン所見(IV型、6歳)
7.治療
1980年代から造血幹細胞移植(骨髄、臍帯血など)が行われ、I型、VI型などでは有効性が明らかになっている。症状が進行する前(2歳以前)に実施することが望ましい。II型に対する移植効果は一定の見解は得られていないが、本邦では軽症型を中心に移植が実施され、一部で有効性が報告されている。中枢神経病変、骨病変、弁膜症に対する効果は部分的であるとされており、適応については移植のリスクも含め慎重な判断が必要である。
酵素補充療法は2003年にI型に対する製剤(一般名ラロニダーゼ、商品名アウドラザイム)が米国で承認されて以来、相次いで開発が進められている。本邦では2006年12月にI型製剤が薬価収載され、II型に対する製剤(一般名イズロサルファーゼ、商品名エラプレース)が2007年に、VI型に対する製剤(一般名ガルサルファーゼ、商品名ナグラザイム)が2008年に薬価収載された。いずれの製剤も尿中ムコ多糖の減少、呼吸機能・歩行距離・肝脾腫・関節可動域・皮膚症状などの改善が認められているが、中枢神経障害、骨病変、弁膜症については効果が得られにくいと考えられている。酵素補充療法はより幅広い患者さんへ適用でき、危険性が低い反面、長期にわたって定期的に点滴を行う必要がある。
研究段階の治療法として、酵素の髄腔内投与、修飾酵素による組織移行の向上、低分子化合物によるムコ多糖の産生抑制、シャペロン療法、遺伝子治療法などが検討されている。
8.参考文献
Neufeld EF, Muenzer J: The Mucopolysaccharidoses. The Metabolic & Molecular Bases of Inherited Disease. 3421-3452. 2001 McGraw-Hill, New York
祐川(早坂)和子、折居忠夫:遺伝性ムコ多糖症.小児内科増刊号 35:483-487, 2003
鈴木康之:Dysostosis multiplex group. 目で見る骨系統疾患 2004 193-196, 2004