
ライソゾーム病に関して(各論)医師向けゴーシェ病
1.概要、欠損酵素
ゴーシェ病は糖脂質が組織に蓄積するスフィンゴリピドーシスのひとつである。 本疾患はグルコセレブロシダーゼ(別名β-グルコシダーゼ)遺伝子変異によりグルコセレブロシダーゼ活性が低下あるいは欠損する。 その結果、生体膜の構成成分であるスフィンゴ脂質の分解過程で基質であるグルコセレブロシドが体中のマクロファージに蓄積し、図1に示すような様々な病態を呈する。
図1
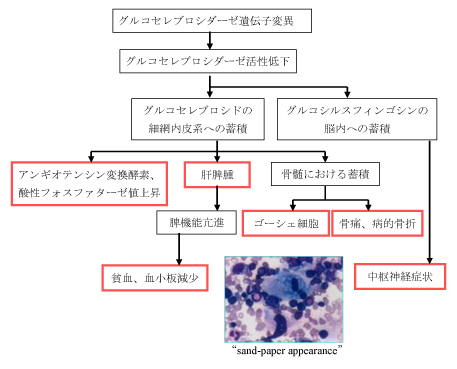
2.遺伝形式
常染色体劣性遺伝形式をとり、発症に性差はない。
3.分類
臨床病型は神経症状の有無およびその重症度により、1型:非神経型、2型:急性神経型、3型:亜急性神経型の3つに分類される(図2)。
| 1型(非神経型) | 2型(急性神経型) | 3型(亜急性神経型) | |
|---|---|---|---|
| 神経症状 | (-) | (+++) | (+) |
| 発症時期 | 幼少期~成人 | 乳児期 | 乳児期~成人 |
| 肝脾腫 | (-)~(+++) | (+) | (+)~(+++) |
| 骨症状 | (-)~(+++) | (-) | (-)~(+++) |
| 予後 | 良好 | 不良 | 良好~不良 |
4.人種差、発症頻度
発症頻度には民族間に大きな差がある。日本人における発症頻度は4~6万人に一人と推定されており、現在まで約150名の患者が同定されている。 病型別にみると3型が最も多い。これに対し、世界的にはアシュケナージ系ユダヤ人では900~1000人に1人、非ユダヤ人では6~10万人に1人が発症するとされている。また、欧米人においては1型が大多数を占める。(図3) この差は遺伝子型の違いに由来する。
図3
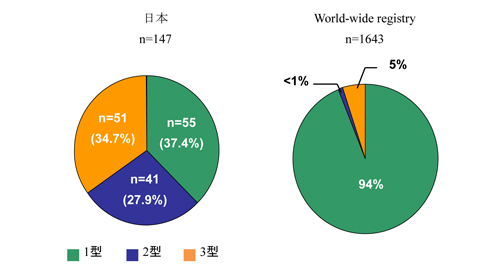
(出典:日本; 自験例、World-wide registry; Charrow J, et al. Arch Intern Med 160, 2000)
5.症状
肝脾腫はどの病型にも共通して認められる。骨折、骨痛、無腐性壊死などの骨症状は重症な1型および3型にみられる。 その他、各臨床型は以下のような特徴をもつ。
- 1型:
- 主症状は貧血、血小板減少、肝脾腫および骨症状で、神経症状は伴わない。 発症時期は幼少期から成人までと幅広く、酵素補充療法による予後は良好である。
- 2型:
- 肝脾腫、肺病変以外にけいれん、後弓反張などの著明な神経症状を伴う。 乳児期までに発症し神経症状が急速に進行して、ほとんどの症例が2~3歳までに死に至る。 新生児期に発症する症例では胎児水腫や魚鱗癬を呈する。
- 3型:
- 神経症状を伴うが2型よりもその程度は軽度で、進行が緩徐である。 神経症状は異常眼球運動、ミオクローヌス、小脳失調、けいれんと多様であるが、その重症度や予後、発症時期により3a、3b、3c型の3つのサブタイプに分類される。 3a型はスウェーデンのNorbotten地方に多い、古典的な3型の病型を呈する。 3b型はより早期に発症し、臓器症状が著明であり、肺高血圧が致死的になることもある。 神経症状は核上性上方注視麻痺のみで1型と誤診される病型である。 3c型では肝脾腫、骨症状は明らかでなく、水頭症、角膜混濁、心弁膜石灰化が主症状である。
6.診断
- 1)生化学的検査:
- 酸性フォスファターゼやアンギオテンシン変換酵素が上昇する。
- 2)画像検査:
- レントゲンで下腿骨のエルレンマイヤーフラスコ変形が、MRIで骨髄のまだら様所見がみられる。
- 3)骨髄検査:
- 骨髄中のゴーシェ細胞の確認。
- 4)酵素活性:
- 皮膚線維芽細胞のグルコセレブロシダーゼ活性測定を行う。 通常、酵素活性は正常の10%以下に低下している。ただし活性の程度と臨床症状の重症度は一致しない。
- 5)遺伝子解析:
- 皮膚線維芽細胞あるいは白血球から抽出したDNAを用いて行われる。 8つのcommon mutation (L444P, F213I, R463C, N370S, 84GG, IVS2+1, D409H, RecNciI)があることがわかっているが、日本人における同定率は約60%である。 一部を除き臨床型と遺伝子型の関連は明らかでない。
診断は酵素活性あるいは遺伝子変異の同定により確定される。これらの検査は東京慈恵会医科大学小児科などの施設で行っている。
7.治療
- 1)酵素補充療法(ERT)
- 本邦ではすべての病型のゴーシェ病に対しERTは保険適応があり実地臨床の場で行われている。 ERTによって、貧血や血小板減少などの血液学的異常および臓器症状は改善されるが、神経症状に対する明らかな効果は乏しいと報告されている。 なお、神経型に対する酵素補充療法に対しヨーロッパ代謝研究グループが勧告治療ガイドラインを提唱している。
- 2)骨髄移植(BMT)
- BMTも保険収載されている。BMTは肝脾腫や血液学的所見の改善だけでなく、 神経症状についてもその進行の停止あるいは改善が期待できるが、 graft versus host diseaseなどの合併症などのリスクがあるため、 実際に行われる症例は限定されている。
- 3)基質合成阻害療法(SRT)
- 欧米ではすでに行われているが、現在国内では1型に対し治験が開始されており、早急な認可が期待される。
- 4)ケミカルシャペロン療法
- 中枢神経症状に対する新しい治療法は以前から検討されているが、 2004年にLinらによりin vivoにおけるN-octyl-β-valienamineを用いたケミカルシャペロン療法が発表された。
- 5)その他の治療法
- 欧米ではERTの大量投与とSRTとの併用を検討した報告があり、神経症状の改善に対し有効である可能性がある。 またグルコセレブロシダーゼを脳に分布させるconvection-enhanced deliveryがラットを使用した実験で成功するなど、新たな治療も開発されている。
1型に関しては主に欧米諸国からその臨床経過や治療効果に関する多くの知見が報告されてきたが、 それらの研究によればERTにより1型ゴーシェ病の予後は多くの症例において良好である。 骨髄移植(BMT)や基質合成阻害療法(SRT)についてもその有効性も報告されている。 一方、2、3型に対しては、現在の治療目標は神経症状の改善を得ることではなく、全身状態やQOLの改善である。